

Esclerosis sistémica: Serie de casos clínicos y revisión integrativa de la evidencia fisiopatológica actual.
Systemic sclerosis: Case series and integrative review of current pathophysiological evidence
[Artículo en español / Article in Spanish]
Vol. 9 Núm. 4. Agosto 2025 - Octubre 2025.
e-ISSN: 2530-5468 - Open Access Journal
DOI: 10.5281/zenodo.17267300
Published under Creative Commons CC BY-NC-ND 4.0

Sanum. vol. 9, número 4 (2025) páginas 100 – 106
AUTORES:
Paola Andrea Yasno Navia MD. Universidad del Cauca, Popayán, Colombia.
Jhan Sebastian Saavedra Torres MD, M. Sc; Esp. MF. Universidad del Cauca, Popayán, Colombia.
Como citar este artículo:
Yasno-Navia, P.A. Saavedra-Torres, J.S. Esclerosis sistémica: serie de casos clínicos y revisión integrativa de la evidencia fisiopatológica actual. SANUM 2025, 9(4) pp 100 – 106 DOI: 10.5281/zenodo.17267300
© Los autores. Publicado por SANUM: Revista Científico-Sanitaria bajo una licencia Creative Commons Atribución-NoComercial-SinDerivadas 4.0 Internacional (CC BY-NC-ND 4.0). ![]()
How to cite this article:
Yasno-Navia, P.A. Saavedra-Torres, J.S. Systemic sclerosis: case series and integrative review of current pathophysiological evidences. SANUM 2025, 9(4) pp 100 – 106 DOI: 10.5281/zenodo.17267300
© The authors. Published by SANUM: Revista Científico-Sanitaria under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND 4.0). ![]()
Tipo de artículo: caso clínico.
Sección: Medicina interna.
Fecha recepción: 12-09-2025
Fecha aceptación: 15-10-2025
Fecha publicación: 31-10-2025
RESUMEN
La esclerosis sistémica (ES) es una conectivopatía heterogénea y potencialmente letal, caracterizada por vasculopatía, autoinmunidad y fibrosis. Describir una serie de cuatro casos y realizar una revisión integrativa de la evidencia fisiopatológica y terapéutica actual. Serie de casos con fenotipado clínico-serológico y revisión narrativa de literatura (1988–2024) en bases indexadas. Se priorizaron guías y estudios de alta calidad. Caso 1: ES difusa con compromiso cutáneo severo y vasculopatía digital. Caso 2: ES difusa con neumopatía intersticial difusa (NID). Caso 3: ES difusa complicada con crisis renal esclerodérmica (CRE). Caso 4: ES limitada con compromiso gastrointestinal esofágico. En todos se documentó relación entre autoanticuerpos (anti-Scl-70, anti-centrómero, anti-RNA pol III) y fenotipo. El desenlace fatal se asoció a NID avanzada, sepsis por úlceras digitales, CRE refractaria y hemorragia digestiva por Barrett/adenocarcinoma. Un factor común fue el abandono terapéutico por barreras geográficas en zonas rurales dispersas. El diagnóstico precoz, la estratificación serológica y el monitoreo protocolizado (PFP/DLCO, TACAR, ecocardiografía) son claves para mejorar supervivencia. El manejo órgano-dirigido (MMF, nintedanib, terapias para HAP, IECA en CRE, IBP/procinéticos) debe integrarse a estrategias que reduzcan inequidades de acceso. Se requieren biomarcadores predictivos y enfoques personalizados que contemplen determinantes sociales de la salud.
PALABRAS CLAVE:
Esclerosis Sistémica;
Autoinmunidad;
Fibrosis;
Autoanticuerpos;
Biomarcadores.
ABSTRACT:
Systemic sclerosis (SSc) is a heterogeneous and potentially life-threatening connective tissue disease, characterized by vasculopathy, autoimmunity, and fibrosis. This study aimed to describe a series of four clinical cases and to conduct an integrative review of current pathophysiological and therapeutic evidence. We present a case series with clinical–serological phenotyping and a narrative literature review (1988–2024) using indexed databases, prioritizing guidelines and high-quality studies. Case 1: diffuse SSc with severe cutaneous involvement and digital vasculopathy. Case 2: diffuse SSc with interstitial lung disease (ILD). Case 3: diffuse SSc complicated by scleroderma renal crisis (SRC). Case 4: limited SSc with esophageal gastrointestinal involvement. In all patients, a clear association was observed between autoantibodies (anti-Scl-70, anti-centromere, anti-RNA polymerase III) and clinical phenotype. Fatal outcomes were linked to advanced ILD, sepsis from chronic digital ulcers, refractory SRC, and gastrointestinal bleeding secondary to Barrett’s esophagus/adenocarcinoma. A common factor was treatment discontinuation due to geographic and social barriers in rural settings. Early diagnosis, serological stratification, and standardized monitoring (PFT/DLCO, HRCT, echocardiography) are essential to improve survival. Organ-specific management (MMF, nintedanib, pulmonary hypertension therapies, ACE inhibitors for SRC, PPIs/prokinetics) should be integrated with strategies aimed at reducing inequities in access to care. Future research must focus on predictive biomarkers and personalized approaches that incorporate social determinants of health.
KEYWORDS:
Scleroderma, Systemic;
Autoimmunity;
Fibrosis;
Autoantibodies;
Biomarkers.
INTRODUCCIÓN:
La esclerosis sistémica (ES), o esclerodermia, es una conectivopatía poco frecuente, heterogénea y potencialmente letal. Su fisiopatología combina vasculopatía no inflamatoria, autoinmunidad y fibrosis progresiva. Se clasifica en forma cutánea limitada (ES-l) y forma cutánea difusa (ES-d) según la extensión del engrosamiento cutáneo (1,2,3). La ES-l —históricamente CREST— se asocia a engrosamiento distal y mayor riesgo de hipertensión arterial pulmonar (HAP); la ES-d compromete piel proximal y tronco, con alta probabilidad de neumopatía intersticial difusa (NID), crisis renal esclerodérmica (CRE) y afectación cardiaca (1,2).
En inmunología clínica, >90% presenta anticuerpos antinucleares (ANA) y ≈60–70% expresa autoanticuerpos específicos, habitualmente mutuamente excluyentes, que orientan el pronóstico: anti-centrómero (fenotipo limitado y HAP), anti-topoisomerasa I / Scl-70 (NID y compromiso cardiaco) y anti-RNA polimerasa III (progresión cutánea acelerada y CRE) (1,2,3).
Esta estratificación debe integrarse a la valoración basal para definir vigilancia: pruebas de función pulmonar (PFP) y capacidad de difusión de CO₂ (DLCO) seriadas; tomografía de alta resolución (TACAR) ante sospecha de NID; ecocardiografía anual para tamizaje de HAP; y control estricto de presión arterial en ES-d reciente. La capilaroscopia ungueal aporta valor diagnóstico y pronóstico (megacapilares, hemorragias y rarefacción) (1,3).
Epidemiológicamente, la ES muestra predominio femenino (~5:1), marcada variabilidad geográfica y curso más agresivo en personas de ascendencia africana. La mortalidad ha cambiado: los inhibidores de la enzima convertidora de angiotensina (IECA) transformaron el pronóstico de la CRE; hoy la enfermedad pulmonar (NID y/o HAP) lidera las causas de muerte. El manejo exige evaluación multisistémica y seguimiento coordinado entre reumatología, neumología, cardiología, nefrología, gastroenterología y dermatología (1,5).
El tratamiento es órgano-dirigido. En NID-ES, micofenolato mofetilo (MMF) es primera línea; ciclofosfamida se reserva para casos seleccionados; nintedanib enlentece el declive funcional y puede coadministrarse con inmunosupresión. En HAP, se emplean inhibidores de fosfodiesterasa-5 (PDE-5i), antagonistas del endotelino (AE) y prostanoides, con escalamiento/combos según riesgo; el cateterismo derecho confirma diagnóstico. En Raynaud/úlceras digitales, los calcioantagonistas dihidropiridínicos son primera línea; puede añadirse sildenafilo, iloprost o bosentán si hay refractariedad. En piel progresiva, metotrexato o MMF son útiles; tocilizumab y rituximab emergen en escenarios tempranos o refractarios (1,2,3).
La CRE exige IECA a dosis máximas toleradas desde el inicio; no se recomienda profilaxis con IECA. En el tubo digestivo, inhibidores de bomba de protones (IBP) en dosis altas y procinéticos, junto con medidas higiénico-dietéticas y manejo de sobrecrecimiento bacteriano (SIBO), son estándar (1,2,4).
Principios operativos: (1) diagnóstico precoz y fenotipado serológico para anticipar complicaciones; (2) monitoreo protocolizado de pulmón, corazón y riñón para intervenir antes de la irreversibilidad; (3) decisiones compartidas ajustadas a riesgo/comorbilidad, evitando corticoides a dosis altas salvo indicaciones muy acotadas. Con esta estrategia, mejora la supervivencia y se minimiza el daño orgánico acumulado (1,2,4).
PRESENTACIÓN DE LOS CASOS:
Caso Clínico No. 1: COMPROMISO CUTÁNEO DIFUSO Y VASCULAR
Una mujer de 35 años, dedicada a la docencia, consultó por fenómeno de Raynaud de cinco años de evolución acompañado de endurecimiento progresivo de la piel en manos, cara y tronco. La rigidez le dificultaba realizar tareas cotidianas y presentaba microstomía con limitación para la higiene oral. Al examen físico se observaron telangiectasias faciales, esclerodactilia y úlceras digitales dolorosas. La capilaroscopia demostró megacapilares y áreas de avasculación, hallazgos compatibles con patrón esclerodérmico activo. Los estudios inmunológicos revelaron anticuerpos ANA positivos y anti-Scl70.
La evolución clínica se explicó por disfunción endotelial persistente y activación anómala de fibroblastos con depósito excesivo de colágeno, responsable del endurecimiento dérmico y de la isquemia digital. El fenómeno de Raynaud severo reflejó la magnitud de la vasculopatía microvascular subyacente.
Se instauró manejo con bloqueadores de canales de calcio y sildenafil para el control vascular, asociado a micofenolato mofetilo con el objetivo de frenar la progresión cutánea. Se indicó fisioterapia para preservar movilidad y acompañamiento odontológico para mitigar complicaciones de la microstomía. Este caso ilustra la importancia del diagnóstico temprano en mujeres jóvenes, ya que el compromiso cutáneo severo suele anticipar afectación visceral y mayor morbimortalidad.
DIAGNÓSTICO: Esclerosis sistémica difusa con compromiso cutáneo severo y vasculopatía asociada.
La paciente falleció por isquemia digital progresiva complicada con sepsis secundaria a infecciones de úlceras cutáneas crónicas, sumada al deterioro multisistémico propio de la vasculopatía esclerodérmica. A pesar de la indicación terapéutica, la paciente abandonó el tratamiento inmunosupresor debido a las dificultades de acceso a controles especializados en su zona rural dispersa, lo que favoreció la evolución hacia complicaciones irreversibles.
Caso Clínico No. 2: COMPROMISO PULMONAR INTERSTICIAL
Un varón de 48 años, agricultor, presentó disnea progresiva en los últimos dos años acompañada de tos seca persistente. Refería fenómeno de Raynaud y artralgias. En la espirometría se documentó patrón restrictivo con capacidad vital forzada reducida al 52% y DLCO en 44%. La tomografía de alta resolución mostró fibrosis pulmonar basal con áreas de vidrio esmerilado. Los estudios inmunológicos confirmaron ANA positivo y anti-Scl70.
La fisiopatología correspondió a remodelado intersticial impulsado por disfunción endotelial y activación de fibroblastos, con pérdida progresiva de la elasticidad pulmonar. Este compromiso constituye la principal causa de mortalidad en la esclerosis sistémica.
El paciente inició tratamiento con micofenolato mofetilo y recibió oxigenoterapia en esfuerzos. Fue remitido a seguimiento periódico con pruebas de función pulmonar seriadas y tomografía de control. Este caso demuestra cómo la afectación pulmonar, silenciosa en etapas iniciales, puede determinar el pronóstico vital si no se aborda de manera temprana.
DIAGNÓSTICO: Esclerosis sistémica difusa con neumopatía intersticial asociada.
El paciente falleció a causa de insuficiencia respiratoria crónica avanzada secundaria a neumopatía intersticial difusa asociada a esclerosis sistémica, refractaria al tratamiento inmunosupresor. La progresión de la fibrosis pulmonar se vio agravada por la descontinuación del manejo farmacológico, ya que el paciente residía en una zona rural de difícil acceso, lo que limitó su adherencia y seguimiento clínico oportuno.
Caso Clínico No. 3: CRISIS RENAL ESCLERODÉRMICA
Una mujer de 50 años, sin antecedentes patológicos relevantes, fue hospitalizada por cefalea intensa, cifras tensionales de 210/110 mmHg y deterioro súbito de la función renal, con creatinina sérica de 3.2 mg/dl. En la anamnesis destacaba historia de fenómeno de Raynaud y engrosamiento cutáneo difuso. Los estudios inmunológicos mostraron ANA positivo y anti-RNA polimerasa III.
La clínica fue altamente sugestiva de crisis renal esclerodérmica, complicación grave caracterizada por vasculopatía obliterante de arteriolas renales. La biopsia no se realizó por riesgo hemorrágico, pero el contexto clínico e inmunológico permitió confirmar el diagnóstico.
Se instauró tratamiento inmediato con inhibidores de la enzima convertidora de angiotensina (captopril), logrando control tensional progresivo y estabilización de la función renal. La paciente ingresó a seguimiento conjunto por reumatología y nefrología. Este caso enfatiza la necesidad de reconocer la crisis renal como emergencia potencialmente letal, en la cual la intervención temprana con IECA mejora de manera significativa la supervivencia.
DIAGNÓSTICO: Esclerosis sistémica difusa complicada con crisis renal esclerodérmica.
La paciente falleció por complicaciones cardiovasculares y renales derivadas de una crisis renal esclerodérmica refractaria, que evolucionó hacia insuficiencia renal terminal y crisis hipertensivas recurrentes. La falta de continuidad en el uso de inhibidores de la ECA estuvo asociada a la imposibilidad de acceso regular a servicios de salud desde su residencia rural dispersa, lo que condicionó la evolución desfavorable.
Caso Clínico No. 4: COMPROMISO GASTROINTESTINAL
Un varón de 55 años consultó por disfagia progresiva y regurgitación nocturna, acompañadas de pérdida de peso no intencionada de 6 kg en seis meses. Presentaba pirosis crónica desde hacía más de tres años. La endoscopia digestiva reveló esofagitis erosiva y dilatación esofágica distal, mientras que la manometría esofágica mostró hipomotilidad marcada. Los estudios inmunológicos fueron positivos para ANA y anticuerpos anticentrómero.
El cuadro clínico correspondió a esclerosis sistémica limitada con compromiso digestivo, derivado de atrofia del músculo liso esofágico y fibrosis progresiva que disminuyen la motilidad y favorecen el reflujo gastroesofágico severo.
Se indicó tratamiento con inhibidores de bomba de protones, procinéticos y recomendaciones dietéticas específicas. El paciente ingresó a vigilancia por gastroenterología para detectar complicaciones como estenosis esofágica o esófago de Barrett. Este caso ejemplifica cómo el compromiso gastrointestinal, frecuente, pero a menudo subestimado, puede impactar de forma importante la calidad de vida y requiere abordaje integral para prevenir complicaciones graves.
DIAGNÓSTICO: Esclerosis sistémica limitada con compromiso gastrointestinal esofágico.
El paciente falleció como consecuencia de hemorragia digestiva masiva asociada a esófago de Barrett complicado con adenocarcinoma esofágico, desarrollado tras años de reflujo gastroesofágico severo en contexto de esclerosis sistémica limitada. A pesar de las recomendaciones médicas, el paciente abandonó el tratamiento con inhibidores de bomba de protones y la vigilancia endoscópica por las barreras geográficas y sociales propias de su entorno rural disperso, lo que facilitó la progresión hacia un desenlace fatal.
DISCUSION:
La presente serie de casos evidencia la heterogeneidad clínica y pronóstica de la esclerosis sistémica (ES), reforzando su carácter de enfermedad multiorgánica en la que el compromiso cutáneo, pulmonar, renal y digestivo pueden coexistir o predominar en distintos momentos evolutivos. Los hallazgos resaltan la importancia de un diagnóstico precoz y de una estratificación serológica y clínica adecuada, dado que las manifestaciones iniciales, como fenómeno de Raynaud, engrosamiento cutáneo o disnea progresiva, suelen anticipar complicaciones graves y determinantes de mortalidad (1,3,5).
La revisión de la literatura confirma que, pese a los avances terapéuticos, la ES mantiene un curso desfavorable en escenarios de acceso limitado a la atención médica, como se evidenció en los pacientes provenientes de zonas rurales dispersas, quienes abandonaron terapias inmunosupresoras o de sostén. Este aspecto pone de relieve no solo la biología agresiva de la enfermedad, sino también los determinantes sociales de la salud como condicionantes del desenlace (1,3,5).
Las limitaciones de este trabajo incluyen el reducido número de casos y la imposibilidad de generalizar los resultados. Asimismo, aunque la revisión bibliográfica integró estudios históricos y actuales, persiste heterogeneidad metodológica en los ensayos clínicos y ausencia de biomarcadores validados para la predicción de evolución (1,2,5).
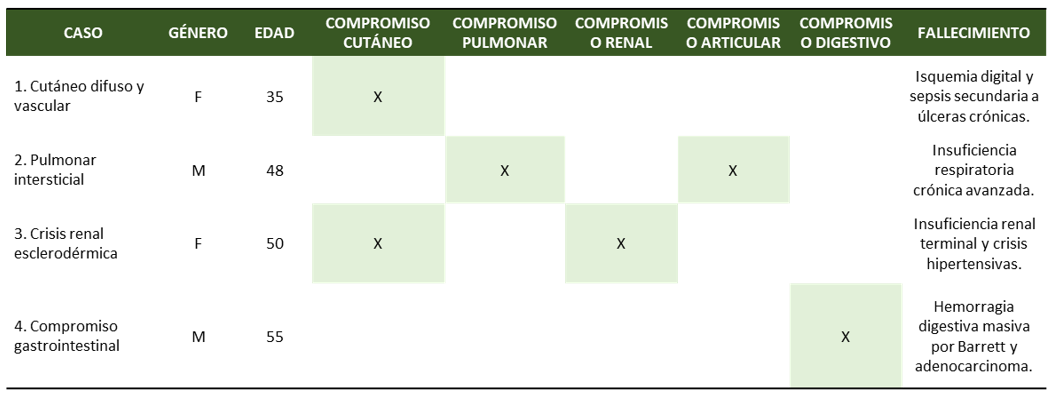
Figura 1. Distribución de las manifestaciones clínicas en cuatro casos de esclerosis sistémica evaluados en la serie. Se evidencia heterogeneidad en el compromiso multiorgánico (cutáneo, pulmonar, renal, articular y digestivo), así como las diferentes causas de mortalidad asociadas (1,2,5). La variabilidad refleja la naturaleza sistémica y progresiva de la enfermedad, donde el abandono terapéutico, condicionado por las limitaciones de acceso en zonas rurales dispersas, representó un factor determinante en la evolución desfavorable de los pacientes. (Fuente: original de los autores).
En términos prácticos, se refuerza la necesidad de implementar protocolos de monitoreo estandarizados y de garantizar continuidad terapéutica en poblaciones vulnerables. A nivel teórico, estos hallazgos estimulan preguntas sobre la interacción entre predisposición genética, microambiente tisular y factores ambientales en la progresión de la ES (1,2,5).
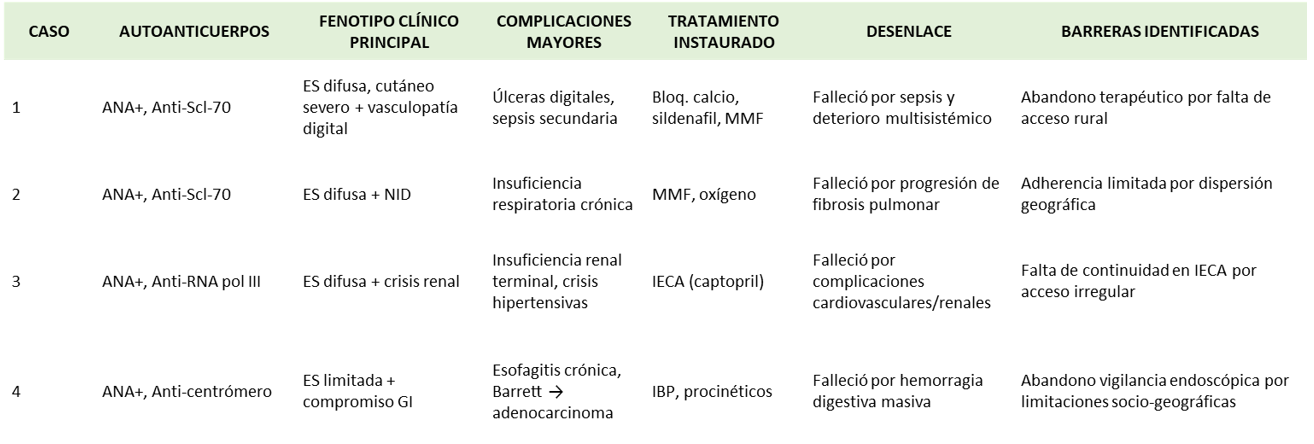
Figura 2. Se integra la relación entre autoanticuerpos, fenotipo clínico, complicaciones y desenlaces fatales en cuatro casos de esclerosis sistémica (ES), resaltando cómo las barreras de acceso en zonas rurales dispersas condicionaron la adherencia terapéutica y el pronóstico (1,2,5). Se evidencia la interacción entre inmunopatología y determinantes sociales de la salud. (Fuente: original de los autores).
DISCUSSION:
This case series highlights the clinical and prognostic heterogeneity of systemic sclerosis (SSc), reinforcing its nature as a multisystemic disease in which cutaneous, pulmonary, renal, and gastrointestinal involvement may coexist or predominate at different stages of evolution. The findings emphasize the importance of early diagnosis and appropriate clinical–serological stratification, as initial manifestations such as Raynaud’s phenomenon, cutaneous thickening, or progressive dyspnea often anticipate severe complications and mortality determinants (1,3,5).
The literature review confirms that, despite therapeutic advances, SSc continues to follow an unfavorable course in settings with limited access to medical care, as observed in patients from rural areas who discontinued immunosuppressive or supportive therapies. This observation underscores not only the aggressive biology of the disease but also the impact of social determinants of health as critical modifiers of outcome (1,3,5).
The limitations of this work include the small number of cases and the inability to generalize the results. Furthermore, although the literature review integrated both historical and current studies, methodological heterogeneity persists among clinical trials, and validated biomarkers for predicting disease progression remain lacking (1,2,5).
From a practical standpoint, the need to implement standardized monitoring protocols and ensure therapeutic continuity in vulnerable populations is reinforced. From a theoretical perspective, these findings raise important questions about the interplay between genetic predisposition, tissue microenvironment, and environmental factors in the progression of SSc (1,2,5).
CONCLUSIÓN:
La esclerosis sistémica representa un reto diagnóstico y terapéutico por su heterogeneidad y potencial letalidad (1,2,4). En esta serie se documentó compromiso cutáneo, pulmonar, renal y digestivo, con desenlaces adversos asociados al abandono terapéutico en contextos de difícil acceso sanitario. Los hallazgos confirman que el diagnóstico temprano, la estratificación serológica y el monitoreo protocolizado son pilares para mejorar la supervivencia. Se reafirma que la mortalidad actual está dominada por la enfermedad pulmonar y que la crisis renal, aunque menos frecuente, constituye una emergencia vital. Futuras investigaciones deberán enfocarse en biomarcadores predictivos y terapias personalizadas, además de estrategias que integren la atención clínica con soluciones a las barreras geográficas y sociales que condicionan la adherencia en pacientes con ES (1,2,5).
CONCLUSION:
Systemic sclerosis remains a diagnostic and therapeutic challenge due to its heterogeneity and potential lethality (1,2,4). In this series, cutaneous, pulmonary, renal, and gastrointestinal involvement was documented, with adverse outcomes largely linked to treatment discontinuation in contexts of limited healthcare access. The findings confirm that early diagnosis, serological stratification, and protocolized monitoring are pivotal to improving survival. Current mortality is driven mainly by pulmonary disease, while renal crisis, though less frequent, persists as a life-threatening emergency. Future research should focus on predictive biomarkers and personalized therapies, alongside strategies that integrate clinical care with solutions to geographic and social barriers that compromise adherence in patients with SSc (1,2,5).
DECLARACIÓN DE TRANSPARENCIA:
La autora principal (defensora del manuscrito) asegura que el manuscrito es un artículo honesto, adecuado y transparente; que ha sido enviado a la revista científica SANUM, que no ha excluido aspectos importantes del caso.
CONSENTIMIENTO INFORMADO:
Se obtuvo el consentimiento informado por escrito de la persona para la publicación de cualquier imagen o dato potencialmente identificable incluido en este artículo. Se obtuvo el consentimiento informado por escrito del participante para la publicación de este informe de caso.
FINANCIACIÓN:
Los autores declaran que no recibieron apoyo financiero para la investigación y/o publicación de este artículo.
CONFLICTOS DE INTERÉS:
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un posible conflicto de intereses.
DECLARACIÓN DE IA GENERATIVA:
Los autores declaran que no se utilizó IA generativa en la creación de este manuscrito.
CONTRIBUCIONES DE LOS AUTORES
P.A.Y.N. y J.S.S.T. han contribuido de manera equitativa a la concepción del estudio, recolección y análisis de la información, redacción del manuscrito, revisión crítica del contenido y aprobación final de la versión a ser publicada. Los autores asumen plena responsabilidad por el contenido y las conclusiones del trabajo.
NOTA DEL EDITOR:
Todas las afirmaciones expresadas en este artículo son exclusivamente del autor y no representan necesariamente las de su organización afiliada, ni las de la editorial, los editores ni los revisores. Ningún producto evaluado en este artículo, ni ninguna afirmación realizada por su fabricante, está garantizada ni respaldada por la editorial.
BIBLIOGRAFÍA:
- González-Serna D, Shi C, Kerick M, Hankinson J, Ding J, McGovern A, Tutino M, Villanueva-Martin G, Ortego-Centeno N, Callejas JL, Martin J, Orozco G. Identification of Mechanisms by Which Genetic Susceptibility Loci Influence Systemic Sclerosis Risk Using Functional Genomics in Primary T Cells and Monocytes. Arthritis Rheumatol. 2023 Jun;75(6):1007-1020. doi: 10.1002/art.42396. Epub 2023 Apr 9. PMID: 36281738; PMCID: PMC10953390.
- Adigun R, Goyal A, Hariz A. Systemic Sclerosis (Scleroderma). 2024 Apr 5. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 28613625.
- Volkmann ER, Andréasson K, Smith V. Systemic sclerosis. Lancet. 2023 Jan 28;401(10373):304-318. doi: 10.1016/S0140-6736(22)01692-0. Epub 2022 Nov 25. PMID: 36442487; PMCID: PMC9892343.
- Lepri G, Di Battista M, Codullo V, Bonomi F, Sulis A, Guiducci S, Della Rossa A. Systemic sclerosis: one year in review 2024. Clin Exp Rheumatol. 2024 Aug;42(8):1517-1528. doi: 10.55563/clinexprheumatol/is29he. Epub 2024 Jul 26. PMID: 39058484.
- Zhu JL, Black SM, Chen HW, Jacobe HT. Emerging treatments for scleroderma/systemic sclerosis. Fac Rev. 2021 May 5;10:43. doi: 10.12703/r/10-43. PMID: 34131653; PMCID: PMC8170563.